仪器社区

带你看文献,只做纯干货
文献精读第27期
文章概述
轴突再生和神经功能恢复在老年人群中极为有限。因此,老年人的神经系统损伤通常会导致严重且长期的残疾。衰老能够引起细胞信号传导的广泛变化,包括代谢、免疫和整体组织稳态的变化,在神经系统生理学和对损伤的反应中发挥了关键作用。目前,我们对衰老依赖的再生失败的分子机制的理解仍然很差,严重阻碍了神经修复疗法的发展,因此,迫切需要确定老化导致再生失败的关键分子和细胞机制。2022年5月13日,英国帝国理工大学的研究人员在《Science》杂志上发表题为“Reversible CD8 T cell neuron cross-talk causes aging-dependent neuronal regenerative decline”的文章,该研究发现了一种衰老依赖的轴突再生衰退的细胞分子机制,依赖于趋化因子CXCL13进行募集和激活的CXCR5+ CD8+ T细胞,在与受损后过表达主要组织相容性复合体Ⅰ类(Major Histocompatibility Complex Ⅰ, MHC Ⅰ)的DRG神经元交流后,会限制轴突再生。此外,作者利用人源化单克隆抗体对CXCL13的拮抗作用,成功逆转衰老小鼠的神经再生衰退并促进了神经系统的恢复,这些结果揭示了一种可用于未来治疗的潜在途径。

核心观点
1、衰老与小鼠SNI损伤后DRG中T细胞活化信号的显著升高有关,这些淋巴活化信号激活了转录因子NF-κB,诱导神经元表达趋化因子CXCL13;
2、SNI损伤后,DRG中受损神经元分泌的CXCL13能够募集CXCR5+ CD8+ T细胞向受损后过表达MHC Ⅰ的神经元附近迁移;
3、CXCR5+ CD8+ T细胞与过表达MHC I的神经元交流激活了Caspase-3,从而损害了pAKT和pS6信号,导致轴突再生失败;
4、通过药理学手段拮抗Caspase-3信号的激活,逆转了老龄动物的轴突再生失败,并恢复了pAKT和pS6的表达;
5、中和CXCL13可阻止CXCR5+ CD8+ T细胞的募集,逆转了衰老依赖的神经再生能力衰退,从而促进SNI后的神经功能恢复。
研究结果分析
1. 衰老导致DRG稳态和神经损伤后适应性免疫基因谱出现广泛变化
在坐骨神经损伤(Sciatic Nerve Injury, SNI)之前(假手术)或之后(SNI),分别对8~10周龄(年轻)和20~22月龄(老年)小鼠的DRG进行RNA-seq检测。与年轻假手术小鼠相比,年轻的SNI小鼠DRG中观察到的神经发育、神经递质、离子转运、G蛋白偶联信号和信号转导等基因表达下调,而在老年的SNI小鼠中未观察到。衰老的DRG中适应性免疫反应相关基因的表达显著富集,特别是在SNI后,T细胞和细胞因子/趋化因子信号显著升高。
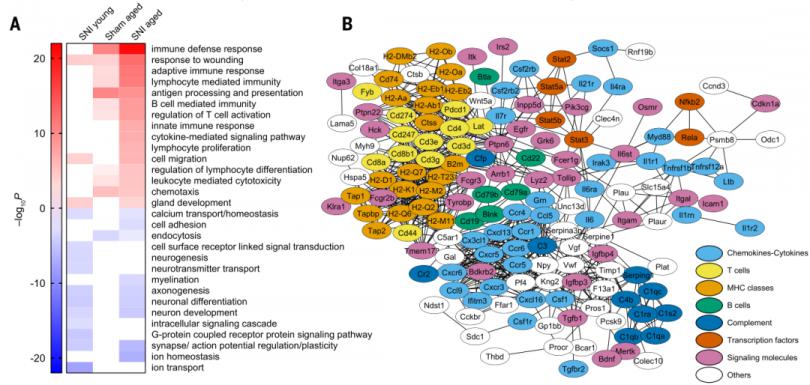
2. 在衰老DRG中,神经元CXCL13和CXCR5+ CD8+ T细胞显著升高
CXCL13是SNI后上调最显著的基因。在衰老的DRG中,CXCL13受体CXCR5的表达也显著增强,提示存在CXCL13-CXCR5信号轴。当衰老的DRG神经元的再生减少时,CXCL13的表达在SNI损伤前和损伤3天后均显著增加。尽管CXCL13在坐骨神经损伤部位也有表达,但是并未观察到衰老相关的变化。在SNI损伤后的第3天,衰老DRG中的CXCL13蛋白水平相对于年轻DRG也有显著增加。

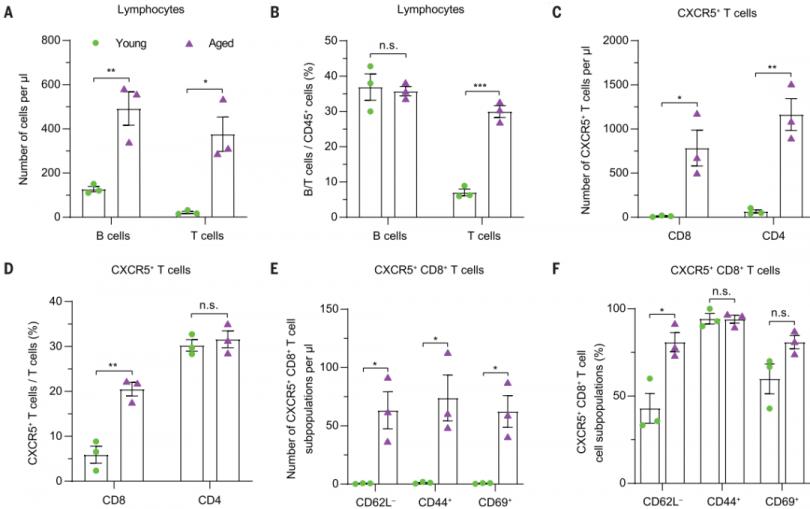
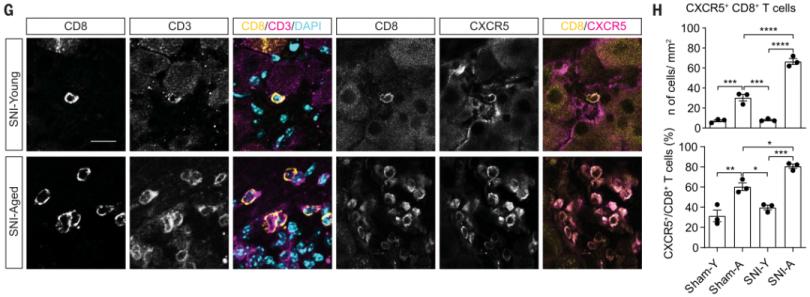
由于CXCL13是CXCR5+ B细胞和T细胞的趋化因子,研究者接下来评估了SNI后3天内年轻或老年小鼠DRG中B细胞和T细胞的定位和免疫表型。衰老DRG中的CXCR5+ B细胞、CXCR5+ CD8+ T细胞、以及CD4+ T细胞明显增多。相当比例的CXCR5+ CD8+ T细胞为CD44+ CD69+ CD62L-,提示它们是效应记忆性T细胞,而只有一个非常少的CD8+ T细胞或CXCR5+ CD8+ T细胞为CD103+和P2RX7+,表明这些细胞是迁移表型而非组织驻留表型。大约一半的CD8+和CXCR5+ CD8+ T细胞也表达CXCR6,表明这些细胞具有滞留和有限再循环的能力。
老年小鼠血液中初始T细胞的总百分率降低,而记忆性CXCR5+ T细胞的百分率显著升高。CD8+ T细胞的数量,包括CXCR5+ CD8+ T细胞,再衰老DRG中显著增加,并且在SNI损伤3天后进一步增加。所有CD8+细胞均为CD68+以及CD3+细胞,这表明它们是T细胞而非巨噬细胞。此外,衰老DRG 中B细胞的数量在SNI损伤后显著增加,但这些细胞DRG实质外,可能是存在于周围小静脉中。因此,衰老与SNI前后神经元的CXCL13表达增加、CXCR5+ T细胞的募集活化、以及T细胞向DRG实质迁移有关。
3. 神经元分泌的CXCL13足以招募CXCR5+ B细胞和T细胞到DRG,并损害轴突再生
接下来,研究者探讨了神经元CXCL13的分泌以及它的过表达是否足以将CXCR5+ B细胞和T细胞募集到DRG中。研究者利用AAV病毒转染培养的DRG神经元,使其过表达CXCL13,并收集培养液用于随后的体外脾细胞迁移实验。过表达CXCL13的培养基增强了CXCR5+ B细胞和T细胞的募集。
为了研究CXCL13的表达是否会影响轴突再生,研究者在体内DRG神经元中过表达CXCL13,并通过干扰素γ(Interferon-γ, IFN-γ)和甘露醇分别诱导MHC Ⅰ的表达和增强血液-DRG屏障的能力,随后立即进行SNI实验。与对照相比,过表达CXCL13的DRG中CXCR5+ B细胞和CXCR5+ CD8+ T细胞均显著增加。过表达CXCL13后坐骨神经轴突再生显著减少。
因此,DRG神经元表达的CXCL13可以招募CXCR5+ B细胞和T细胞,并可以抑制轴突再生,部分复制了衰老表型。


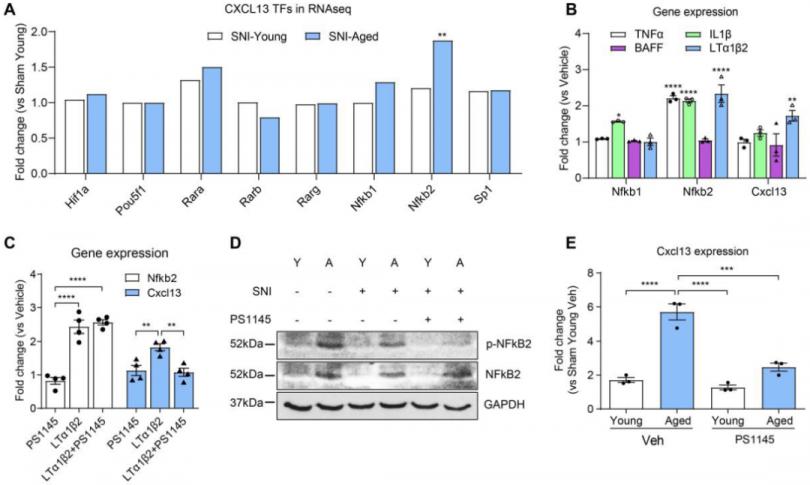
4.衰老DRG中CXCL13的表达依赖NF-κB的磷酸化
为了确定转录因子(Transcription Factors, TFs)是否参与驱动衰老依赖的CXCL13表达,研究者分析了促进CXCL13表达的TFs在衰老DRG中的表达情况。在已知的与CXCL13启动子结合的TFs中,只有NF-κB2在衰老DRG中显著上调。随后,研究者用典型的NF-κB激动剂(TNF-α或IL-1β)和非典型的NF-κB激活剂(BAFF或LTα1β2)来处理培养的DRG神经元24小时。TNF-α和IL-1β能够上调NF-κB1和NF-κB2的表达,但不能上调CXCL13的表达,而BAFF既不影响NF-κB的表达也不影响CXCL13的表达。然而,非典型的NF-κB激活剂LTα1β2能够显著增强NF-κB2和CXCL13的表达,这种作用可以被NF-κB激酶IKK的抑制剂PS1145减弱。NF-κB的表达和磷酸化均随衰老而增加,但相对于对照,PS1145抑制了NF-κB的磷酸化。因此,衰老诱导的CXCL13的表达在PS1145中也显著降低。此外,对磷酸化的NF-κB进行免疫组化染色显示,磷酸化的NF-κB在SNI前和3天后在DRG神经元内定位,并衰老的DRG神经元中显著增加。因此,年龄相关的CXCL13表达是由NF-κB驱动的。
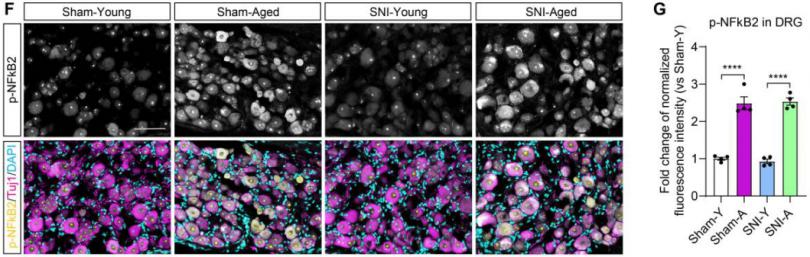
5. CD8+ T细胞和神经元中MHC I的表达是衰老依赖的神经损伤再生衰退所必需的
接下来,研究者探讨了CD8+ T细胞、CD4+ T细胞或B细胞是否参与了老化DRG神经元轴突再生的衰退。在老年小鼠SNI前1周,用CD8α单克隆抗体来耗竭CD8+ T细胞,小鼠SNI手术后,其轴突再生能力得到显著改善,DRG中CD8+ T细胞显著减少。相比之下,CD4+ T细胞或B细胞的耗竭并没有改变老年小鼠SNI后衰老相关的轴突再生衰退。这些数据表明CD8+ T细胞在老年动物SNI后的轴突再生能力下降中起着重要作用。
RNA-seq显示,SNI后MHC I的表达与年龄相关。与年轻对照相比,衰老DRG中MHC I的表达在SNI前后显著增。随后,研究者探讨了CD8+ T细胞是否是为限制MHC I表达的DRG神经元轴突生长所必需。利用IFN-γ能够诱导MHC I在DRG神经元中表达,TCR转基因的OT-I CD8+ T细胞在卵清蛋白肽(OVA257–264)存在时能够有效的降低这些表达MHC I的神经元轴突生长、同时增加Cleaved Caspase-3水平,但没有增加其它凋亡特征。因此,DRG神经元的轴突生长减少依赖神经元MHC I的表达和OT-I抗原特异性的CD8+ T细胞。
接下来,研究者探讨了MHC I依赖性的抗原递呈在衰老小鼠的轴突再生衰退中是否必要。为此,研究者利用AAV-GAr-rtTA病毒在衰老小鼠DRG中条件表达编码的甘氨酸/丙氨酸多肽序列GAr,该序列可抑制MHC I抗原递呈,从而避开CD8+ T细胞的免疫反应,然后进行坐骨神经损伤。与对照相比,AAV-GAr-rtTA感染后神经元的MHC I和Cleaved Caspase-3表达显著降低,坐骨神经轴突再生显著增强。因此,SNI后衰老相关的神经再生衰退依赖于CD8+ T细胞和神经元MHC I的表达。
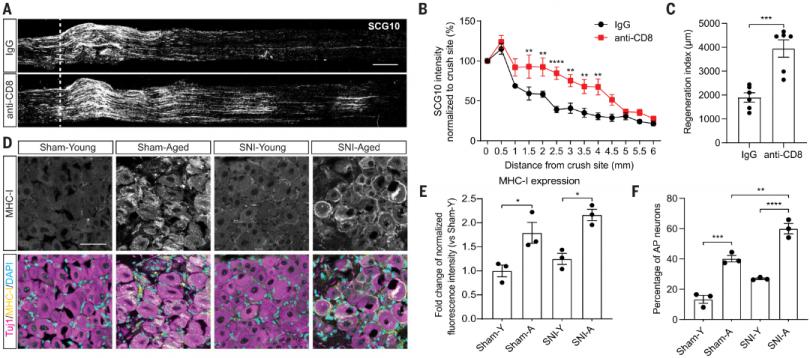
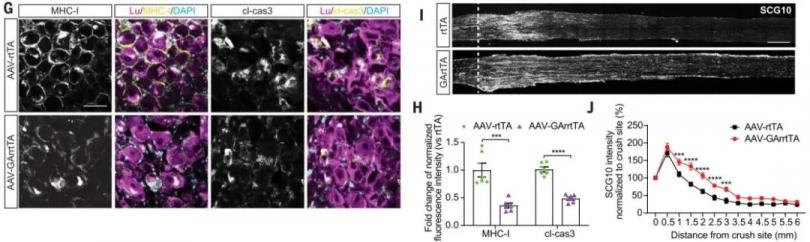
6. CD8+ T细胞依赖的神经元Caspase-3激活抑制了pAKT和pS6信号,这与衰老相关的轴突再生失败有关
为了阐述CD8+ T细胞依赖性的衰老相关的再生衰退的分子信号。在SNI后3天,老年小鼠DRG神经元中Cleaved Caspase-3的表达显著增加,而其中与凋亡相关的DNA片段缺失。SNI损伤后,老年DRG神经元中Perforin和Granzyme B的表达显著增加。激活Caspase 3可导致AKT截断和AKT磷酸化水平降低,进而降低S6磷酸化水平。pAKT和pS6与轴突再生相关,并且通常会随着年龄的增长而受损。事实上,在SNI后第3天,衰老DRG中pAKT和pS6水平下降,然而,这种效应在CD8+ T细胞耗竭后被显著逆转。CD8+ T细胞耗竭显著增强了DRG神经元中的pAKT和pS6的表达,同时减少了Cleaved Caspase-3、Perforin、和Granzyme B的表达。最后,利用Z-DEVD-FMK抑制Caspase-3能够显著增加pAKT和pS6,并增加坐骨神经的轴突再生。因此,在SNI后衰老DRG神经元中,CD8+ T细胞依赖的Caspase-3激活会抑制pAKT和pS6,而抑制Caspase-3的激活可促进神经损伤后轴突再生。
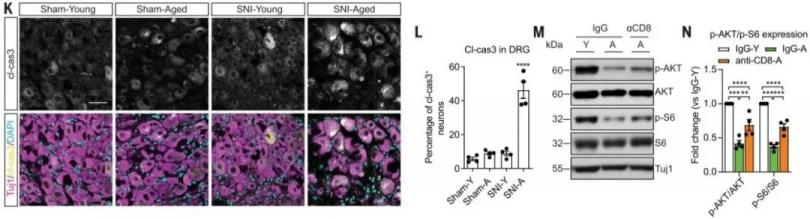
7. CXCR5+ CD8+ T细胞驱动SNI后衰老依赖的轴突再生衰退
接下来,研究者试图确定CXCR5+ CD8+ T细胞是否直接导致SNI后轴突再生能力下降。CXCR5+ CD8+ T细胞被分离并移植到缺乏B细胞和T细胞的年轻或年老的OT-I Rag2-/-小鼠体内。在细胞移植之前,老年OT-I Rag2-/-小鼠无论CD8+ T细胞是否耗竭,其神经再生能力相对于野生型小鼠显著增加。因此,内生的OT-I CD8 + T细胞对神经再生没有影响。
然后,研究者探究了CXCR5+ CD8+ T细胞的移植是否足以限制老年小鼠SNI后坐骨神经的再生。将CXCR5+ CD8+ T细胞注射到年轻或年老的OT-I Rag2-/-小鼠体内。CXCR5+ CD8+ T细胞充分地浸润到年老而非年轻小鼠DRG中,相反,CXCR5+ CD8+ T细胞在年轻小鼠的脾|脏中积累更多。CXCR5+ CD8+ T细胞的这些迁移模式与DRG和脾|脏中不同的CXCL13水平相关。此外,CXCR5+ CD8+ T细胞在老年小鼠的血液中增加。最后,CXCR5+ CD8+ T细胞移植导致老年小鼠坐骨神经损伤后轴突再生显著降低。CXCR5+ CD8+ T细胞移植导致老年小鼠DRG神经元中Perforin的表达显著增加、pS6的表达显著下降,而在年轻小鼠的DRG中没有发现这些变化。
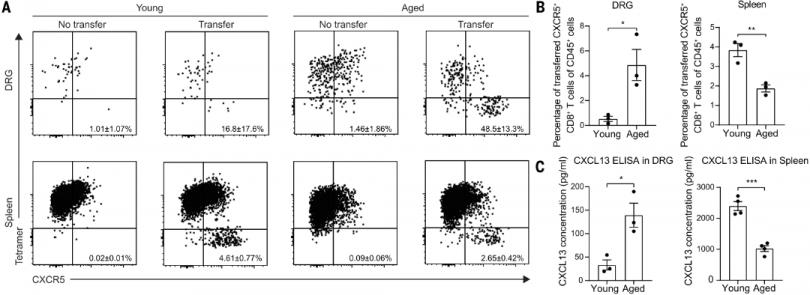

8. CXCL13拮抗剂阻断CXCR5+ CD8+ T细胞的募集,挽救了SNI伤后衰老依赖的轴突再生衰退
已经证明CXCR5+ CD8+ T细胞的募集会导致轴突再生失败,作者探究了这些细胞是如何募集的。将分选的野生型CD8+ T细胞或Cxcr5-/- CD8+ T细胞移植到OT-I Rag2-/-小鼠体内,小鼠在SNI手术前用对照免疫球蛋白G或CXCL13单克隆抗体处理。DRG中Cxcr5-/- CD8+ T细胞的募集明显低于野生型CD8+ T细胞。CXCL13单克隆抗体显著降低了CXCR5+ CD8+ T细胞向DRG以及脾|脏的迁移。CXCL13拮抗显著增强了移植野生型CD8+ T细胞而非移植Cxcr5-/- CD8+ T细胞小鼠的SNI后轴突再生能力。因此,在衰老DRG中,CD8+ T细胞的迁移和限制坐骨神经再生需要依赖CXCL13作用于其受体CXCR5。


9. CXCL13中和逆转了衰老依赖的再生衰退,促进了SNI后的神经功能恢复
最后,作者研究了CXCL13中和是否能促进衰老动物的轴突再生、表皮神经支配以及神经功能恢复。给予CXCL13单克隆抗体显著提高了老年小鼠体外培养DRG神经元的轴突再生,减少了CXCR5+ T细胞和B细胞的募集,阻止了衰老依赖的坐骨神经再生失败以及SNI后DRG中CD8+ T细胞的浸润。此外,作者测试了拮抗CXCL13对神经功能恢复的影响。CXCL13中和显著加速了老年小鼠感觉功能的恢复。CXCL13中和能显著促进老年小鼠后爪皮肤神经再生。因此,CXCL13中和能促进老年动物轴突再生、皮肤神经再生和神经功能恢复。

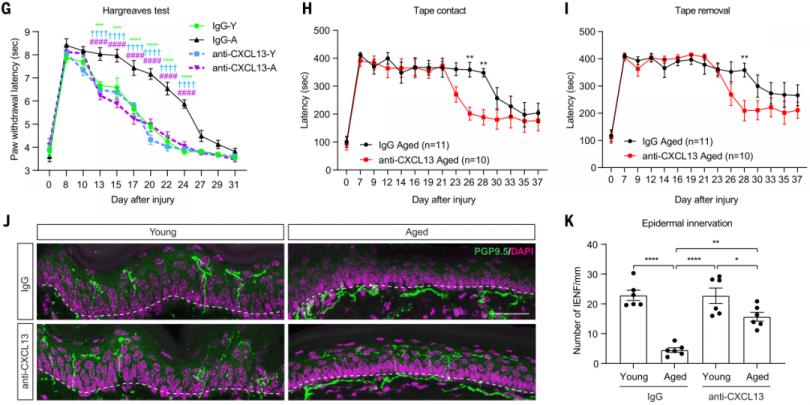
总结
该研究表明,衰老通过一种CXCL13依赖机制,调控CXCR5+ CD8+ T细胞进入DRG,导致轴突损伤后DRG神经元轴突的再生失败。再生失败是CXCL13依赖的CXCR5+ CD8+ T细胞的DRG募集的结果,这些细胞通过DRG神经元表面的MHC I与神经元交流,从而驱动轴突再生抑制信号。因此,该研究的结果提示,CXCL13中和可能可以用于治疗老年人的神经损伤。更普遍的讲,这些结果描述了特定年龄的神经再生机制,表明这些有针对性的干预措施需要考虑损伤或疾病开始时的年龄。

亮点研究方法
这项工作阐述了衰老依赖的神经再生失败的细胞分子机制。研究用到了动物手术造模、组织病理检测、细胞分子检测、给药以及行为学评估等实验技术。瑞沃德深耕生命科学研究领域20年,一直致力于为客户提供可信赖的解决方案和服务,可提供的该研究中涉及的动物手术造模、行为学评估、光遗传学、电生理记录、组织病理检测、给药以及分子检测等实验的完整解决方案。截至目前,瑞沃德产品及服务覆盖海内外 100 多个国家和地区,客户涵盖700+医院,1000+科研院所,6000+高等院校,已助力科研人员发表SCI文章14500+,获得行业广泛认可。

原文链接:
https://www.science.org/doi/10.1126/science.abd5926
 啮齿类动物的外周神经损伤如何评估?
啮齿类动物的外周神经损伤如何评估?

 评论
评论
