仪器社区
具有独特选择性的C18 键合相:
1.有保证的可重现性
2.的键合相稳定性
3.疏水和五氟苯基“混合模式”的相互作用
改善色谱分离度
色谱分离的目标是在Z短的时间内获得目标组分的足够分离度(Rs)。
1.5的分离度可以实现基线分离,然而对于可以在实验室之间易于转换的耐用、可重法而言,理想的分离度是1.8-2.0。
分离度方程告诉我们什么变量可以影响分离度:
 | Rs = 目标峰之间的分离度 N = 柱效- 由理论塔板数测定 α= 选择性- 两峰的保留值比率(k值) k =保留因子- 洗脱峰所需的柱体积数 |
增加分离度Rs可以通过增加N、α或k来实现。
然而,如图1所示,可以看出,增加N或k以改善Rs的回报率快速下降。
例如,Rs仅随着N平方根的增加而增加。
可以通过增加柱长或降低柱填充材料的粒径或两者的某种组合来增加N。
无论哪种方式,系统背压随着N的增加而增加,因此通过增加N实现令人满意的分离,其“成本”可能是极高的压力。
同样,增加保留值(k值)将会增加Rs,但回报率也快速下降。
将k增加至超过10通常是Rs与分析时间之间的不利权衡,因为只有Rs的边际收益随着保留时间的增加而实现。
该效应的图形表示参见下述图1。
图1 N、α和k对分离度(Rs)的影响
对于典型的分离,其中:N = 10,000, k = 4, α= 1.1

增加N、α或k可以提高分离度(Rs)。
然而,从这些图中可以看出,N或k的提高都会迅速降低回报率。
另一方面,提高选择性(α)则没有这个问题,因此其成为开发分离方法时的Z佳优化变量。
增加α可以增加Rs,但不同于N和k,不会受回报率下降的约束。
α的变化对压力没有影响,对分离时间的影响也是微乎其微的(参见图2)。
因此,在开发分离方法时,α是Z重要的变数。
优化α可以使您在所有目标峰之间达到满意的分离度,同时保持系统背压和分离时间在可接受范围内。
改善色谱分离度- 选择性或柱效?
选择性(α)由流动相、温度和固定相化学物质控制。大多数方法开发策略将探索所有这些色谱变量。
如果使用“标准”3μm C18相没有达到足够的分离度,推荐优化分离的色谱选择性而不是分离柱效,如下述实例所示。
通过简单地将固定相化学物质(即色谱柱)改变为具有替代色谱选择性的固定相化学物质,易于在标准HPLC系统上获得所需分离度,而无需昂贵的UHPLC仪器。
另外,也可以避免复杂的流动相组分、升高的温度和侵蚀性pH条件。
图2利用选择性实现快速、高分离度的分离
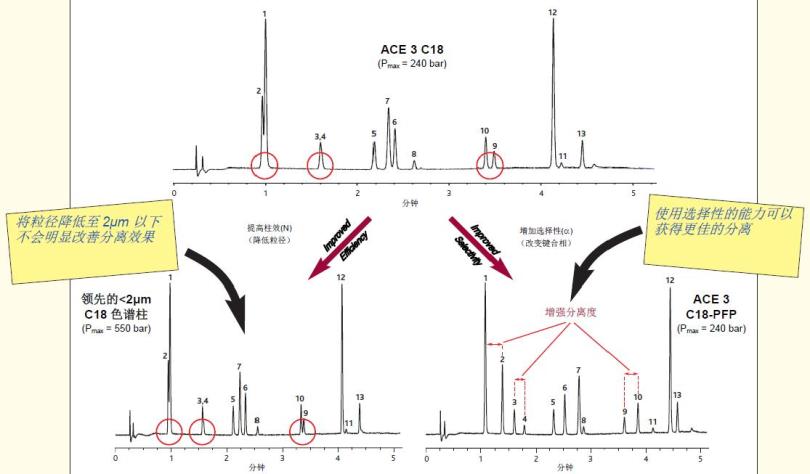
样品:1)对乙酰氨基酚2)氢氯噻嗪3)甲基苯基亚砜4)甲基苯基砜5)阿司匹林6)非那西丁7)1,3-二硝基苯
8)1,2,4-三甲氧基苯9)苯甲酸乙酯10)尼美舒利11)布洛芬12)吲哚美辛13)甲芬那酸
色谱柱尺寸:50 x 2.1 mm 流速:0.60 ml/min 温度:40°C 检测:UV, 254 nm 流动相:A = 5 mM甲酸(溶于水中)以及B = 5 mM(溶于甲醇中),梯度= 在5分钟内3- B
比较数据不代表所有应用。
在保持C18键合相的同时,将粒径从3μm减小至2μm以下,并不能显著改善分离效果,另外也会导致压力明显增加。
ACE C18-PFP色谱柱为3个关键对提供了更佳的选择性(α),因此与2μm以下的C18色谱柱相比,其可以提供的分离效果,即使2μm以下的色谱柱可以提供更高的柱效。
与使用具有高塔板数和高压的色谱柱尝试进行峰分离所获得的结果相比,利用选择性的能力可以获得更佳的分离效果。
 干货丨这么多气相毛细管色谱柱,哪个型号规格最合适?
干货丨这么多气相毛细管色谱柱,哪个型号规格最合适?
 气液相色谱柱保存小贴士:“我”也需要过个舒适年
气液相色谱柱保存小贴士:“我”也需要过个舒适年
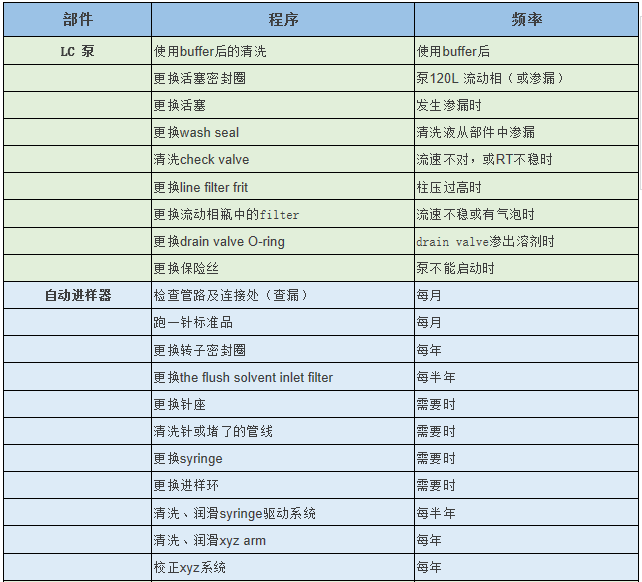 实验室用户必备:Thermo Fisher液相色谱仪、色谱柱如何维护?
实验室用户必备:Thermo Fisher液相色谱仪、色谱柱如何维护?

 评论
评论
