物理吸附之应用篇
2024-08-01371. 物理吸附分析仪(比表面和孔隙度分析仪)的工作原理是什么?
由于没有工具对比表面进行直接测量,人们就根据物理吸附的特点,以已知分子截面积的气体分子作为探针,创造一定条件,使气体分子覆盖于被测样品的整个表面(吸附),通过被吸附的分子数目乘以分子截面积即认为是样品的比表面积。比表面积的测量包括能够到达表面的全部气体,无论外部还是内部。物理吸附一般是弱的可逆吸附,因此固体必须被冷却到气体的沸点温度,并且选择一种理论方法从单分子覆盖中计算表面积。比表面和孔隙度分析仪器就是创造相应的条件,实现复杂计算的这样一种仪器。
2. 比表面积值是测出来的吗?
比表面积值不是测出来的,是计算出来的。我们测量的是样品的吸附等温线,然后根据样品的特性,选择恰当的理论模型计算出样品的比表面积。所以,比表面的测定过程实际是一个分析过程。由于不同的人对样品的认知可能不同,对同一组吸附等温线的实验数据分析可能会报告不同的比表面积结果。 因此,在“测定”比表面的时候,要牢记这是一个“分析”过程。22. BET 就是比表面吗?计算比表面积的方法有多少种?
BET 法只是比表面分析方法中的一种理论。Langmuir 第一次揭示了吸附的本质,其方法是单分子层吸附理论,适合于仅有微孔的样品分析。
BET 理论发表于 1938 年,其正式名称是多分子层吸附理论,是对 Langmuir 理论修正。BET是该理论的三个提出者姓氏的首字母缩写。由于 BET 法适合大部分样品,目前成为zui流行的比表面分析方法。但 BET 法并不适用于所有样品,因此按介孔材料的分析方法分析微孔材料时,由物理吸附分析仪自动生成的 BET 比表面值是错误的。ISO9277-2010 和 IUPAC 都对含微孔材料的 BET比表面分析方法及判断 BET 结果的方法做出了规定。不同的理论模型给出的计算结果是不同的,所以要根据理论模型的假设条件,选择zui适合样品性质的理论模型。大多数理论模型是根据发明人的名字或缩写命名的,能计算出比表面的理论模型
包括 Langmuir,BET,BJH,DR 和 NLDFT。NLDFT 是非定域密度泛函理论。研究表明,NLDFT 计算出的比表面值zui接近真实值,并且该理论适用于微孔和介孔材料。
3. 通过物理吸附测定比表面的原则是什么?
常用的吸附气体是氮气,它已经成为比表面分析的标准吸附物质。这是因为高纯度的氮气很容易得到;另外,液氮作为zui合适的冷却剂也很容易得到;其三,氮气与大多数固体表面相互作用的强度比较大;zui后,氮气分子在77.35K时的截面面积为0.162nm 2,这个在BET计算中必须用到的数值已经被广泛接受。在传统的容量法技术中,小于整数的相对压力是通过造成部分真空条件来实现的。在已知的固定体积里,用精确的高精度压力传感器监控因吸附过程引起的压力变化情况。需要测得在不同相对压力下一系列的气体吸附量。通常,测定仪器在相对压力范围0.025 和0.30 之间至少采集3 个数据点。实验测定的数据以成对数值的方式进行记录:以在标准温度和压力(STP)下的体积 (VSTP) 表示气体吸附量,其对应的是相对压力 (P/Po)。根据这些数据绘制的图就称为吸附等温线。
4. 在物理吸附分析中,应该至少了解哪些重要术语?
在比表面积计算和仪器参数设置中,应该会接触到以下术语或参数:
(1) 阿伏加德罗常数:6.022x10 23
(2) BET:这是三个人的名字缩写,他们分别是:S. Brunauer,P.Emmet 和E.Teller。他们是用
多层气体吸附理论计算比表面积的发明者。
(3) 截面面积(Cross-sectionalArea):单个被吸附的气体分子所占有的面积。
(4) 摩尔体积:一摩尔气体所占有的体积。等于在标准温压下的22,414cc(22.414 升)
(5) 摩尔(无量纲):含有阿伏加德罗常数个数的原子或者分子的一种物质的量。
(6) 单分子层:由下标m表示,它的意义是厚度仅仅为单个分子厚度的一层被吸附的气体。
(7) 相对压力P/Po:绝对压力P与饱和蒸汽压力之比。其值在0和1之间。
(8) 饱和蒸汽压力Po:在给定温度下,一种气体液化时的压力。
(9) 标准温压体积:在标准温度为0℃(273.15K) 和一个标准大气压下,一定数量的气体所占有的体积。
5. 比表面和孔径分析为什么常用氮气?用其它气体可以吗?
如前所述,气体分子是作为吸附探针来分析比表面的,所以它应该满足以下应用条件:
1)气体分子相对惰性,保证不与吸附剂发生化学作用;
2)为了使足够气体吸附到固体表面,测量时固体必须冷却,通常冷却到吸附气体的沸点,因此要求冷却剂相对容易得到;
3)符合或满足理想气体方程的使用条件。
在恒定低温下测量气体的吸附和脱附曲线,所使用的气体是那些在固体表面形成物理吸附的气体,尤其是在77.4K时的氮气、77.4K或87.3K时的氩气、或195K和273.15K时的二氧化碳。因为氮气非常便宜,所以作为被吸附物质得到广泛应用。由于气体分子尺寸各异,可以进入的孔也各不相同,因此测量温度不同,得出的结果可能不同。
由于氮气不是完全的惰性气体,与孔壁可以发生四极矩作用,IUPAC 于 2015 年正式建议,氮气不适合微孔样品的分析,应该采用 87K 下的氩气作为吸附气体。
6. 比表面和孔径分析为什么要用液氮?不用可以吗?
如果用氮气作为被吸附气体,固体样品在分析时就需要被冷却到液氮的沸点温度 (77.35K)。液氮是相对容易得到的价格低廉的实验材料,因此,我们要用液氮获取样品所需要的温度。但需要注意的是,只有纯的液氮才能达到这个温度,而不纯的液氮因温度偏高会造成计算误差,不能使用。另外,暴露于空气中的液氮会冷凝空气,造成液氮纯度下降。所以,实验后剩余的液氮应弃之不用,而不能倒回液氮储罐从而造成贮存液氮的纯度下降。
如果不使用液氮,我们可以采用机械制冷的方式使样品端处于 77.35K。目前,商用Cryocooler 低温恒温系统可在 20K 到 320K 之间设置样品分析温度,极大地方便了实验设计。
7. 如何判断液氮不纯?
因为氮气占空气中的比例为 78%,其饱和蒸汽压约为大气压加 10。出现以下情况,说明液氮明显不纯:环境大气压为 760mmHg,但测出的氮气饱和蒸汽压大于 790mmHg;液氮颜色发蓝,说明其中含有液氧;测出的氮气饱和蒸汽压为 750mmHg,但环境大气压仅有 700mmHg,与当时的大气压比明显偏高; 仪器的液位传感器“失灵”,探测不到液位。这说明液位温度可能高于传感器设置的温度响应范围;
分析过程中线性很好,但偏离常规值很多。
8. 在进行物理吸附分析前,为什么要对样品进行脱气处理?
在进行气体吸附实验之前,固体表面必须清除污染物,如水和油。大多数情况下,表面清洁(脱气)过程是将固体样品置于一玻璃样品管中,然后在真空下加热。右图展示了预处理后的固体颗粒表面,它含有裂纹和不同尺寸和形状的孔。

9. 如何选择样品的脱气温度?
系统温度越高,分子扩散运动越快,因此脱气效果越好。通常仪器配备的脱气站加热温度可达 400 ℃,但是选择脱气温度的首 要原则是不破坏样品结构。一般来说,氧化铝、二氧化硅这一类氧化物的安全脱气温度可达 350 ℃;大部分碳材料和碳酸钙的安全脱气温度在 300 ℃左右;而水合物则需要低得多的脱气温度。对于有机化合物,也可以通过脱气站进行预处理,但是大部分有机化合物的软化温度和玻璃化温度较低,因此必须提前加以确认。例如在医药领域常用的硬脂酸镁,美国药典(USP)规定的脱气温度为 40 ℃。如果脱气温度设置过高,会导致样品结构的不可逆变化,例如烧结会降低样品的比表面积,分解会提高样品的比表面积。但是如果为了保险,脱气温度设置过低,就可能使样品表面处理不完全,导致分析结果偏小。因此在不确定脱气温度的情况下,建议使用化学手册,如 theHandbook ofChemistry and
Physics(CRC,BocaRaton,Florida),以及各标准组织发布的标准方法,如 ASTM,作为相关参考。脱气温度的选择不能高于固体的熔点或玻璃的相变点, 建议不要超过熔点温度的一半。当然,如果条件许可,使用热分析仪能够zui精确地得到适合的脱气温度。一般而言,脱气温度应当是热重曲线上平台段的温度。
10如何确定样品的脱气时间 ?
与脱气温度对应的是脱气时间。脱气时间越长,样品预处理效果越好。脱气时间的选择与样品孔道的复杂程度有关。一般来说,孔道越复杂,微孔含量越高,脱气时间越长;选择的脱气温度越低,样品所需要的脱气时间也就越长。可以通过在相同脱气温度下,分析样品的 BET 结果变化来确定脱气时间。如果在不同的脱气时间(2 小时,4 小时和 6 小时)得到的 BET 结果相同,肯定选择脱气时间zui短的;如果变化不大,则需要选择折衷的方案;如果 BET 结果随脱气时间延长不断变大,说明孔道复杂,深层次有因氢键结合的吸附水分子,暴露了被堵塞的孔道及面积。对于一般样品,IUPAC 推荐脱气时间不少于 6 小时,而那些需要低温脱气的样品则需要长得多的脱气时间。对一些微孔样品,脱气时间甚至需要在 12 小时以上。但是作为特例,美国药典(USP)规定硬脂酸镁的脱气时间就仅为 2 小时。
由于脱气温度、脱气时间以及脱气真空度都与比表面积值有关,所以 BET 结果存在误差是不可避免的。所以,测样时需要固定样品处理条件进行相对比较。与文献值比较时,也要注意文献上的样品预处理和分析条件。
11 样品脱气时,应该选择真空脱气还是流动脱气?两种方法各有什么特点?
流动脱气一般是用于比表面快速分析的,它对于除去表面大量弱结合的吸附水非常好, 但对在孔道中吸附的水,只有经长时间吹扫使之扩散至表面,才能被带出。
真空脱气对于除去表面大量弱结合的吸附水是不好的, 因为水会在泵中扩散,导致泵的抽力下降。 但对孔中吸附的水,不需要经很长时间就能扩散至表面,继而被带出。所以,对于含水量较高的样品,应先在烘箱中烘烤过夜,再上真空脱气站,以保护真空泵。对于真空脱气来说,其对样品清洁能力明显优于流动脱气(见下图),但同时需要考虑的是真空度不同,脱气效率是明显不同的。对于含有超微孔样品,深层次的吸附水分子因氢键结合可以堵塞孔道,它们必须经过分子泵脱气才能清除, 即脱气站真空度必须达到与分析站同样的真空度。5埃分子筛脱气处理后的程序升温脱附(TPD)5 埃分子筛脱气处理后的程序升温脱附
曲线:
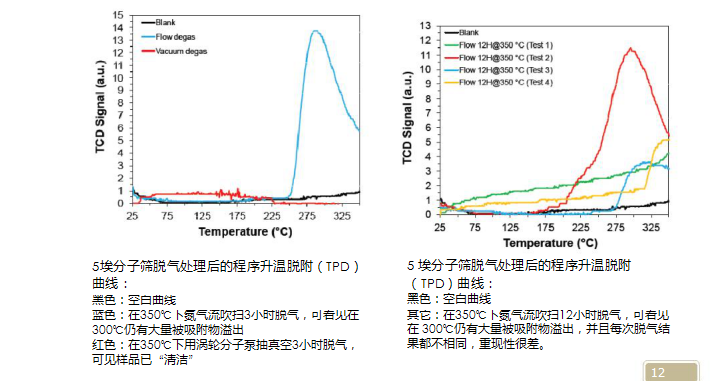
12. 对于亲水性超微孔样品脱气,应该有什么要求?
分子泵的真空脱气方法。这样,样品可以在完全的无油系统中实现脱气对于吸附测定往往起始于相对压力(P/P 0)10-7的微孔材料,特别推荐通过低真空隔膜泵加上涡轮.
亲水微孔样品的脱气是极具挑战性的,因为从窄微孔去除以前吸附的水非常困难。所以,高温(350℃)和长的脱气时间(通常不低于 8 小时)是必需的。对于一些沸石分子筛样品,还需要特殊的加热程序,即在低于 100℃的温度下,可以缓慢除去大部分预吸附的水。其脱气温度是逐步增加的,直到zui终脱气温度为止。这样做是为了避免由于表面张力的影响和蒸汽的水热蚀变作用(hydrothermalalteration),造成样品的电位结构遭到破坏。
zui近的 ISO9277:2010 标准《固体气体吸附比表面积的测定 -BET 法》要求:对于敏感的样品,建议采用压力控制的加热方式(见下图)。此过程包括在真空脱气的条件下,伴随着多孔材料的气体压力的变化,改变加热速率。当从样品表面解吸下来的物质使压力超过一个固定的限制 P(通常大约 7 到 10Pa),升温即停止并温度保持恒定,直到压力低于极限。然后,系统继续升温。
此方法对避免微孔材料的结构变化特别适用,因为较快的加热速率导致大量的吸附水集中汽化,从而破坏脆弱的微孔结构。另外,该方法对防止超细粉末材料因孔道中水蒸汽或其他蒸汽的释放导致的扬析是非常安全的。

13. 脱气后应该回填什么气体并卸载?
zui好选择吸附气体(氮气)作为回填气体,以防止或尽量减少气体浮力所带来的称重误差。当样品管中回填氦气时,与回填空气或氮气相比,样品管的重量会少很多,大约每毫升样品池体积能引入 1 毫克的误差。如果称样量<50 毫克时,这个称重误差是非常显著的。
14. 物理吸附测量的实验技术都有哪些?
物理吸附分析主要测量的是在一定温度下,样品吸附量与压力的关系,即吸附等温曲线。吸附量作为压力的函数可以由体积测量法(容量法)和重量分析法实现。
1)重量分析法是由一个灵敏的微量天平和一个压力传感器构成,可以直接测量吸附量,但是需要做浮力修正(而浮力是无法直接测量的)。重量分析法在以室温为中心的不太大的温度范围内进行时,是一种很方便的研究方法。在重量法中,吸附质不能与温度调节装置直接相连,所以无论在低温或高温,都不容易控制和测量吸附质的真实温度。因此,在液氮温度下(77.35K)或液氩温度(87.27K)下测量氮气、氩气和氪气吸附主要依靠体积测量法。
2)体积测量法即真空容量法,是基于被校准过的体积和压力,利用总气量守恒实现的。利用进入样品管的总气体量和自由空间中的气体量的差值计算出吸附量。体积测量法和重量分析法都需要被测量吸附反应发生在静态和准平衡状态下。在准平衡状态下,被吸附气体以一定的低速率连续地进入样品管,而脱附曲线是通过压力的连续降低获得的。相关准静态平衡过程的zui难点是我们需要达到每时每刻的令人满意的平衡状态。为了检测这样的平衡状态,应该反复利用缓慢的气体释放速率(投气)进行分析。如果在两个不同的气体速率下获得相同的数据,就可以确认分析结果的正确性。这种方法的主要优势在于它能够达到真的平衡状态,并可得到极高分辨率的吸附等温线。
3)连续流动法:和准静态平衡方法相反,这是载气(氦气)和吸附气体(如,氮气)的混合气流连续通过放有样品的流化床的方法。样品吸附氮气会引起气体组成的改变。热导检测器(TCD)可以监控这一变化,并由此计算出吸附量。这个方法仍然广泛用于单点比表面积的快速分析。
15. 什么是自由空间?什么是死体积?它对测量灵敏度有什么影响?
真空体积法进行物理吸附实验是在一个密闭空间进行的。样品管阀门以上的歧管体积和样品管阀门以下的体积共同组成了静态容量法物理吸附测量中所需的系统体积,在后者的这个空间中(即样品管的空间中),除了样品所占据的体积,剩余的空间就是自由空间(free space),其所占据的体积叫死体积(voidvolume)。自由空间是系统中吸附质分子传递、扩散的区域,如果要精确计算样品的物理吸附量,死体积值是准确采集数据的基础。因为真空体积法的测量基础是压力,吸附量的计算基础是理想气体状态方程,所以吸附质气体在扩散过程中压力差越大,则气体绝对量计算越准确。系统死体积越小,对压力变化的灵敏度越高,吸附量计算越准确。换句话说,在同样的条件下,系统死体积越小,则仪器测量精度越高。
16. 测定自由空间的死体积有哪些方法?
在测定吸附等温线之前或之后,应该测定死体积。根据 ISO15901 标准,测定死体积有两种方法:
1)测量法:在测定温度下,采用氦气进行体积校准。这是经典的死体积测定方法,精度zui高。
其应用前提是基于以下两个假设:
i.氦气不被吸附剂材料吸附或吸收;
ii.氦气不能渗入吸附物质(如氮气)不能进入的区域。
2) 校准曲线法:将死体积的测定从吸附测定中分离开来,事先用吸附气体测空管进行空白实验,然后保存待用(NOVA 方式)。例如,在环境温度下先将空样品管的体积用氮气测定,随后,再在与吸附测定相同的实验条件下(温度和相对压力范围相同)用该空管进行一次空白实验。得到的校准曲线实质上代表了多点自由空间的检测。通过输入样品密度(即骨架密度)对样品体积进行必要的校正,或在环境温度下,吸附分析开始前,用氮气测定比重(如果氮气在室温的吸附效应可以忽略不计)。这种方法不仅适用于比表面积分析和介孔等温吸附线的测定,还可以节约氦气,并将管路材质对吸附气的吸附校正包括在内。针对特定样品管的空白曲线可以多次使用,因此省略了每个样品都需要用氦气测死体积的步骤,缩短了分析时间,是一种快速测定比表面或吸附曲线的方法。IUPAC 在 2015 年的报告中还特别指出,校准曲线法对包含极其狭窄的微孔的沸石和活性炭的吸附剂是有利的,也就是说,NOVA 方式测定死体积也适用于沸石分子筛和活性炭的微孔分析,对MOF 材料的适用性也得到了实验支持。
17. 微孔孔径与气体压力有什么关系?
在微孔中,孔壁间的相互作用势能是相互重叠的,因此微孔内的物理吸附比在较宽的孔内或外表面的物理吸附要强(见右图)。于是,在非常低的相对压力(<0.01)下微孔被顺序充填。也就是说,孔径与压力有对应关系,随着压力从高真空逐渐增加,气体分子总是先填充zui小的孔(c),再填充较大的孔(b),然后是更大一点的孔(a),以此类推。在无限长狭缝微孔中表面与孔内流体间相互作用的势能随微孔宽度变化关系的放大示意图(横坐标:孔壁间距;纵坐标:相互作用势能)


18. 静态容量法物理吸附分析仪一般由哪些部分组成?
静态容量法的分析仪器多种多样,为了不同的应用目的设计有不同的特点,但都包含以下基本要素(如下图):一个真空泵、一个或多个气源、一个连接样品管的金属或玻璃歧管、一个冷却剂杜瓦、一个样品管、一个饱和压力测定管、一个压力测量装置(压力传感器)。歧管的体积需要进行校准。需要具有记录歧管温度的手段。可以使用各种尺寸的样品管,体积一般为 10~20cm 3。为尽可能减小误差,样品上方的自由空间应尽可能减小,可以通过在样品管的颈部放入玻璃棒(填充棒)来减小死体积。

19. 物理吸附分析仪对气体纯度有什么要求?
因为吸附气体是用来评估吸附剂表面积和孔径的,氦气是用来测定死体积的,所以根据ISO15901 的要求,这些气体的纯度必须在 99.99%以上,但 IUPAC 在 2015 年的zui新报告中指出,
吸附气体的纯度不得低于 99.999%。
20. 为什么要称量样品质量(称重)?称样量多大为好?
比表面积是单位质量的表面积,所以必须在脱气后和分析前对样品管中的样品用减重法进行称重计量。对于氮气吸附测定,我们要考虑样品在样品管中的总表面积,也就是比表面积 X 样品质量。
样品量以总表面积达到5~200m 2之间为好。用天平计量出来的是质量,不是重量。为方便说明,此处混用这两个概念。测试前应对仪器精度(可测量的zui小总表面积)及样品的表面积有个大概的估计,以确定所需样品量。小于仪器精度,测试结果相对误差则较大。当样品有很高的比表面积时,称样量较少,这时称量过程可能带来较大的误差。称样量的判断原则是:
1)尽量减少天平的称重误差;
2)样品管中的样品要有足够的吸附量,其引起的压力变化应远大于压力平衡所允许的公差(Tolerence);
3)样品管中的样品吸附量也不能过大,否则吸附平衡时间太长,导致实验时间过长;
4)如果仅需要比表面积测量,则称样量应使样品管中的总表面至少在1-5m 2 间;
5)如果是测定吸脱附等温线,在样品管中的总表面积应至少为15-20m 2。
对于氮气吸附,有关称重的经验如下:
尽可能称重到100mg以上,以减少称重误差
如果比表面大于1000:
称重0.05-0.08g
如果比表面大于10,小于1000: 称重0.1-0.5g
如果比表面小于1:
称重需要在1g以上,甚至到5g以上
为确保测量精度,分析后应重新称量样品的质量。如果分析后的质量不等于脱气后、分析前的
初始质量,应采用分析后的质量进行重新计算。
21. 样品管都有哪些规格?样品管和填充棒的选择原则是什么?
一般厂家都能提供 6mm、9mm 和 12mm 管径的多种规格样品管。管径越细,死体积就越小,测量精度也就越高,但装填样品时比较困难。所以,要根据样品情况,权衡利弊,酌情使用。
减小冷自由空间是所有仪器设计制造人员的共识。所以,选择样品管时,都遵从“尽量使用填充棒、尽可能细的样品管颈、尽可能小的样品舱”原则。
除此之外,还需综合考虑如下因素:
1) 样品形态的影响:对于粉末样品,尤其是低密度的粉末样品,如活性碳等,在抽真空的过程中粉末扬起会引起分析结果不准确,如果粉末沾染到 O 形圈将造成系统漏气,一旦粉末进入系统歧管还将引起更加难以修复的系统污染。因此,对于这类样品推荐使用管颈相对较粗、样品舱相对较大的管子,并且不推荐使用填充棒。而对于大颗粒、高密度样品,如金属、某些分子筛等,受抽真空力影响较小,不会引起系统污染,因此选择样品管就可以直接遵从首 要原则,“尽量使用填充棒、尽可能细的样品管颈、尽可能小的样品舱”。
2)分析类型的影响:对于微孔材料的孔径分析,由于实验起始压力相对低(通常从相对压力10-7/10-6 区段起始),在低温下分子扩散速率较慢,加之气体非理想性对数据采集的影响较大,因此推荐不使用填充棒,以减少实验误差。对于介孔段的孔径分析以及比表面积测试,由于气体非理想性对数据采集的影响极小,因此使用填充棒反倒是可以提高实验结果的准确性。
3)样品比表面积的影响:对于小比表面样品,在试验过程中所需样品量较大,通常需要几克甚至十几克。这种情况下,为保证实验的准确性,应注意的是样品量不要超过样品舱(直管、小球或大球)总体积的 2/3。此外,若小比表面样品还具备 1)中所提到的密度小的特性,那么也不
推荐使用填充棒。
样品管的选择经验有如下参考:
9mm 样品管是zui常用的样品管,适合大部分样品;
标准样品管用于颗粒样品及常规比表面分析;
大球样品管用于粉末样品及低表面样品分析;
6mm 样品管对于高精度的微孔分析是非常必要的。
22. 什么是歧管?它对仪器测量精度有何影响?
歧管(manifold)是物理吸附分析仪中连接进气端口、真空系统、压力传感器和样品管等的多支路管路系统。歧管体积是计算物理吸附初始进气量的依据之一。这部分体积固化在仪器内部,可通过校正得到精确数值。另一方面,吸附质气体在扩散过程中压力差越大,则气体绝对量计算越准确。因此,歧管体积越小,则仪器精度越高。
23. 为什么要记录歧管温度?歧管温度控制对测量精度有什么影响?
在理想气体方程中,体积、压力均为温度的函数,因此,准确的系统温度也是吸附量准确计算的一个基础。通常系统温度是通过与歧管相连的温度传感器实时记录的。目前市售的大部分仪器大多使用精度±0.1 ℃的温度传感器,均可满足实验精度的要求。但是必须指出的是,zui新的仪器设计趋势是所谓“高分辨微孔分析”的技术,该类型仪器均采用 0.1torr 压力传感器采集低压区数据,以使在高真空区域(相对压力<10 -6)的数据分辨率和稳定性更高。但是,该类型传感器对温度变化更为灵敏,因此,为了获得数据的高稳定性,需要特别配置更为稳定的系统温度,例如采用系统加热的方式,保持歧管恒温在 50 ℃,避免温度波动。
如果是静态高压吸附系统,歧管温度波动±0.5 ℃,就会造成吸附量计算的明显误差(如±0.3mol@CO2),因此要求对歧管温度的控温精度在±0.1 ℃以内。
压力传感器作为静态容量法的基本计量单元,应该自身都有电子陶瓷恒温系统。如果选用没有恒温装置的压力传感器,虽然成本较低,但压力测量精度也会极低,就没有可能测量 10nm 以上的较大介孔分布。
24. 在分析过程开始前,为什么要除掉氦气?
在用氦气测量死体积时,是基于氦气不吸附的假设。但事实上,物理吸附是非特异性吸附,对任何气体都存在吸附,因此,某些材料,特别是微孔材料会吸附较多的氦气,其影响无法忽略不计,
也就是存在氦污染。氦污染的典型现象是吸附等温线在 P/P0<10-5以下时出现“S”线形。因此,对于这种情况,应该关心死体积测定后,是否经历了除氦过程,再进行等温线测定;或在测定吸附等温线之后,对其进行修正。IUPAC 在 2015 年的报告中指出:zui近的研究已经证实,具有极窄微孔的纳米多孔固体可以在液氮温度下吸附无法忽略的氦气量(氦截留)。如果在分析之前不除去被截留的氦气,可以显著影响
在超低压范围的吸附等温线的形状。因此,建议在继续分析之前,应当至少将样品放在室温下使氦气溢出后,将其脱气。 氦气作为单原子分子,直径只有 0.26nm,远小于氮气分子的截面积,可以进入氮气不可能进入的极细孔道。研究表明,用氦气在液氦温度下分析活性炭纤维的 BET 比表面
比在液氮温度下用氮气表征,其 BET 比表面积值增加 1/3。
25. 什么是冷自由空间?什么是暖自由空间?冷暖自由空间的相对大小有什么意义?
在分析过程中,样品管是部分浸没在冷浴(如液氮)中的,因此总自由空间是由冷自由空间和暖自由空间两部分组成的。其中浸没于液氮液位下的部分称为 冷自由空间(冷域,cold-zone),在液位以上处于室温环境部分称为 暖自由空间(暖域,warm-zone)。自由空间是系统中吸附质分子传递、扩散的区域。液氮温度(77K)下同体积所包含的分子数是室温 300K 的 4 倍,可以说在整个自由空间中冷自由空间对死体积的贡献远大于暖自由空间。冷自由空间越小,或者暖自由空间中所含气体分子数越多,压力测量也就越准确。
26. 为什么要进行液氮或液氩的液位控制?控制液位都有哪些方法?
在一个敞开的杜瓦中,制冷剂如液氮和液氩是挥发的。因此,样品管颈的制冷剂液位是不断降低的,从而造成冷域和暖域体积的连续变化。为避免系统自由空间受冷浴温度及冷浴液位的影响,
在测量中的关键就是保持样品管颈与制冷剂液位的相对恒定。一般要求,至少浸没样品 20mm,并保持液面恒定,波动不超过 1~2mm。在实际操作中,有两种不同的方式达到上述目的:
(1) RTD 实时反馈伺服方式, 即利用包括液位传感器和自动电梯在内的实时反馈伺服系统调整冷浴液位并保持冷自由空间zui小化。实验证明,用液位传感器控制浸于液氮中的样品池液位,得到的样品管中的氮气压力(大约 295 毫米汞柱)与时间的函数关系见右图。液氮液位的任何变化都会造成管内气压的变化。压力恒定的结果清楚地说明,液位控制伺服反馈系统补偿了液氮蒸发的损失,极好地控制了样品管中的死体积(zui小的“冷域”和zui大的“暖域”)保持恒定。
(2) 夹套方式,即用高分子多孔材料包裹样品管颈,通过毛细管蒸腾作用保持液位,也就是用较大冷自由空间换取冷浴液位高度的恒定。
27. 什么是饱和蒸汽压?为什么要测饱和蒸汽压?
在液体(或者固体)的表面存在着该物质的蒸汽,这些蒸汽对液体(或固体)表面产生的压强叫作该液体(或固体)的蒸汽压。在一定温度下,与同种物质的液态(或固态)处于平衡状态的蒸汽所产生的压强就是饱和蒸汽压。
当样品管浸于制冷剂(如液氮)中,样品管中的纯吸附物质(氮气)呈现的饱和平衡蒸汽压用P0表示。液态纯物质蒸汽所具有的压力为其饱和蒸汽压力时,汽液两相即达到了相平衡。所以,真空饱和吸附,吸附压力不可能大过吸附物质的饱和蒸汽压,即相对压力(P/P 0)不可能大于 1。 饱和蒸汽压是吸附物质的一个重要性质,它的大小取决于物质的本性和温度,与吸附层厚度、
孔填充压力以及孔中的毛细管凝聚有关。 饱和蒸汽压越大,表示该物质越容易挥发。只有得到准确的气体饱和蒸汽压,通过吸附量与相对压力 P/P0关系的精确表征才能进行准确的孔径及比表面积分析。
28. 如何测量饱和蒸汽压?
饱和蒸汽压的大小与温度相关。有多种实验方法可以用于计算物理吸附过程中的饱和蒸汽压。但是准确度zui高的方法是在物理吸附实验过程中在独立的 P0管中连续测量饱和蒸汽压。通常吸附等温线都是在液氮(77.35K)或液氩(87.27K )温度下测量,液氮、液氩放置于杜瓦瓶中,保持常压。此时液体温度不仅与压力,更与液体纯度相关。水蒸汽、氧气以及空气中的其它气体组分均可影响液体纯度,当液体纯度降低则液体温度也会随之升高,温度升高幅度 0.1~0.2K 可导致饱和蒸汽压上升 10~20torr。在物理吸附过程中,当相对压力为 0.95 时,饱和蒸汽压的误差达 5 torr 时,会导致孔径计算的近 10 %误差。因此在物理吸附过程中准确、实时地测量饱和蒸汽压是非常重要的。
具有独立饱和蒸汽压传感器的仪器,能够实时监测 P0的变化,而对实验过程不产生干扰。但是,若相对压力中的饱和蒸汽压并非取自该点平衡时刻的饱和蒸汽压,则测量的精度依然会有很大折扣,这对微孔材料的微孔分布分析有着重要意义。
29. 物理吸附分析系统的进气模式都有哪些?各有什么特点?
由于物理吸附分析系统测定的基础数据是平衡吸附量与压力的关系,因此我们必须设定一个量值,而测定另一个量值。这样,就产生了两种进气模式:
(1) 定投气量模式(设定纵坐标,测量横坐标):
由仪器采集压力信息的方法称之为“定投气量方式”。该方法对于仪器硬件及固件设计的要求较低,是各个生产厂家广泛使用的方法。该方法的一个亮点是可以扩展进行吸附动力学的相关研究以及低温反应的相关研究,但对于常规的微孔孔径分布分析,定投气量方式存在如下不确定性: 如果投气量设置过小,得到的等温线固然细节丰富,但是却与实验所花时间呈反比。如果投气量设置偏大,等温线上的部分信息就会丢失。投气量设置偏大,可以缩短测试时间,但并没有达到真正的吸附平衡,造成吸附等温线向右“漂移”,导致微孔分析的误差(见图 49-1)。
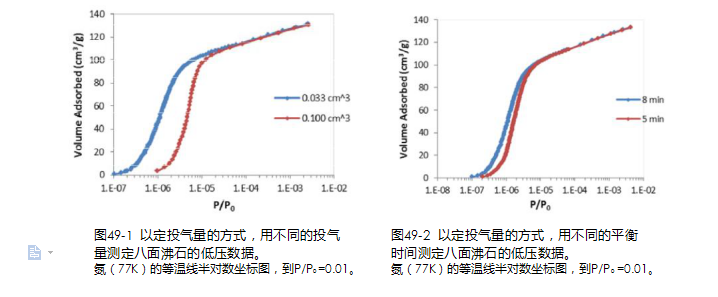
图49-1 以定投气量的方式,用不同的投气
图49-2 以定投气量的方式,用不同的平衡
量测定八面沸石的低压数据。
氮(77K)的等温线半对数坐标图,到P/P0 =0.01。时间测定八面沸石的低压数据。氮(77K)的等温线半对数坐标图,到P/P0 =0.01。IUPAC 在 2015 年的报告中指出:太短的平衡时间会导致未平衡的数据生成,等温吸附线移向过高的相对压力。因为在窄微孔中的平衡往往是非常慢的,未平衡往往是在等温线的极低相对压力区域内容易发生的问题(见图 49-2)。
(2) 定压力方式(设定横坐标,测量纵坐标):
由仪器采集并计算饱和吸附量的方法称之为“定压力方式”,该方法zui大的优点是:由仪器内置程序计算各定义压力下的吸附量,这种方法对于吸附量未知的样品可以既快又准地得到吸附等温线,
尤其对于未知的微孔样品。快速、准确地测量与数据的准确性同样具有重要实践意义。但是,定压力方式对内置程序设计要求极高,尤其是对于微孔定压力测量(实验起始相对压力需达到 10-7~10-5量级),必须同时考虑饱和蒸汽压、系统体积、样品量等信息,具有其复杂性。不正确的“定压力方式”宏命令编程设计很容易导致等温线测量的偏差。
30. 吸附平衡条件是如何设置的?
在静态容量法物理吸附实验中,所谓吸附平衡是在一定的扩散时间内,体系中气体压力变化始终在允许误差范围内的状态。它与投气方式共同组成了物理吸附仪器测量准确度中zui核心的环节。若平衡时间不够,则所测得的样品吸附量或脱附量小于达到平衡状态的量,而且前一点的不完全平衡还会影响到后面点的测定。例如,测定吸附曲线时,在较低相对压力没有完成的吸附量将在较高的压力点被吸附,这导致等温吸附线向高压方向位移。由于同样的影响,脱附曲线则向低压方向位移,形成加宽的回滞环,或者产生不存在的回滞环。 对于微孔测量,由于其孔径较小,需要的平衡时间相应增加。
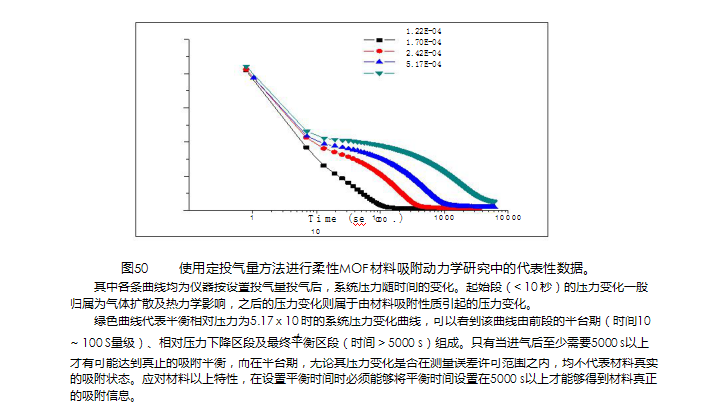
使用定投气量方法进行柔性MOF材料吸附动力学研究中的代表性数据。其中各条曲线均为仪器按设置投气量投气后,系统压力随时间的变化。起始段(< 10 秒)的压力变化一般归属为气体扩散及热力学影响,之后的压力变化则属于由材料吸附性质引起的压力变化。 绿色曲线代表平衡相对压力为5.17 x 10 时的系统压力变化曲线,可以看到该曲线由前段的平台期(时间10 -4~ 100 S量级)、相对压力下降区段及zui终平衡区段(时间 > 5000 s)组成。只有当进气后至少需要5000 s以上才有可能达到真正的吸附平衡,而在平台期,无论其压力变化是否在测量误差许可范围之内,均不代表材料真实的吸附状态。应对材料以上特性,在设置平衡时间时必须能够将平衡时间设置在5000 s以上才能够得到材料真正的吸附信息。 如果说平衡时间(Equilibriumtime)规定的是达到平衡的zui低时间要求,那么平衡压力误差
(Tolerance)则是用于认定达到平衡时允许压力变化范围的参数。这两个参数共同决定了吸附平衡条件。随着各种特色新材料的快速涌现,吸附平衡条件设置必须具有足够的灵活性以适应不同类型材料分析的需求。例如,对于柔性MOF材料(也有人称之为会呼吸的材料),由于其孔道结构变化需要相当长的时间,在实验平衡条件设置时,必须能够针对具体材料的孔道结构变化时间设定仪器的平衡时间(见图50)。
因此,能否进行灵活的吸附平衡条件设置就成了衡量物理吸附仪器测量准确度的一个重要标准,
-
- 贝士德 全自动高温高压气体吸附仪BSD-PH
- 品牌:贝士德仪器
- 型号:BSD-PH

-
- 贝士德 腐蚀性气体吸附分析仪BSD-PMC
- 品牌:贝士德仪器
- 型号:BSD-PMC

-
- 贝士德 多组分吸附穿透曲线分析仪BSD-MAB
- 品牌:贝士德仪器
- 型号:BSD-MAB

-
- 贝士德 全自动化学吸附仪BSD-C200
- 品牌:贝士德仪器
- 型号:BSD-C200

